100
%
JoinMap ® 5
JoinMap ® 5
Neogen Informatics is an authorised and preferred reseller of Kyazma softwares in India.
JoinMap is an MS-Windows ® program for the calculation of genetic linkage maps in experimental populations of diploid species. The software can deal with the common types of experimental population, including the full-sib family (F1) of a cross between two individuals of an outbreeding species. It provides high quality tools that allow detailed study of the experimental data. The intuitive user interface invites to a better exploration of the data. It is easy to perform various diagnostical tests, both before and after the actual map calculation. Subsequently, any locus or individual with possibly erroneous observations can be excluded from the calculations by a simple mouse-click, after which a new and improved map may be calculated.
Salient features:
- MS Windows based.
- Many experimental population types:
- BC1 - first generation backcross;
- F2
- RIx - recombinant inbred lines family.
- DH1, DH - family of F1-derived doubled haploids.
- DH2 - family of F2-derived doubled haploids.
- HAP1, HAP - family of haploids.
- BCpxFy - advanced backcross inbred lines family.
- IMxFy - advanced intermated inbred lines family.
- CP - outbreeder full-sib family.
- Input in plain text files with a flexible layout;
- Input also by pasting marker data copied from MS-Excel.
- Also imports MAPMAKER raw data format (data types: f2 intercross, f2 backcross, ri self).
- Easily check for genotype coding errors in Dataset node.
- Several diagnostics, before and after the actual map calculation:
- Test segregation distortion.
- Check similarity of loci.
- Check similarity of individuals.
- Calculate genotype probabilities conditional on map and flanking genotypes to discover double recombinations
- Test heterogeneity of recombination estimates between different populations.
- Powerful determination of linkage groups.
- Four criteria to study the linkage group formation: independence LOD, independende test P-value, linkage LOD, recombination frequency.
- Use existing maps (of multiple groups) or existing groupings to create the linkage groups of a new population.
- This is very useful when employing markers with known map positions in new populations, and also when expanding a map with an additional set of markers.
- Use the so-called strongest cross link information to verify assignments of markers to groups.
- Automatic determination of linkage phases for outbreeder full-sib family.
- Very fast computation of high density maps with the ML mapping algorithm according to Jansen, et al, 2001. Constructing dense genetic linkage maps. TAG 102: 1113-1122, based on Monte Carlo (MC) Maximum Likelihood (ML).
- Has since v4.1 the ability to use the ML mapping algorithm also on populations of type CP (outbreeder full-sib family); method according to: Van Ooijen, J.W. (2011). Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genetics Research (2011) 93, 5, 343-349.
- Since v5 the Gibbs sampler for the estimation of multipoint recombination frequencies is replaced by the deterministic forward-backward EM algorithm, which is much faster and more accurate.
- The new ML mapping algorithm and the original regression mapping algorithm of JoinMap are available side by side.
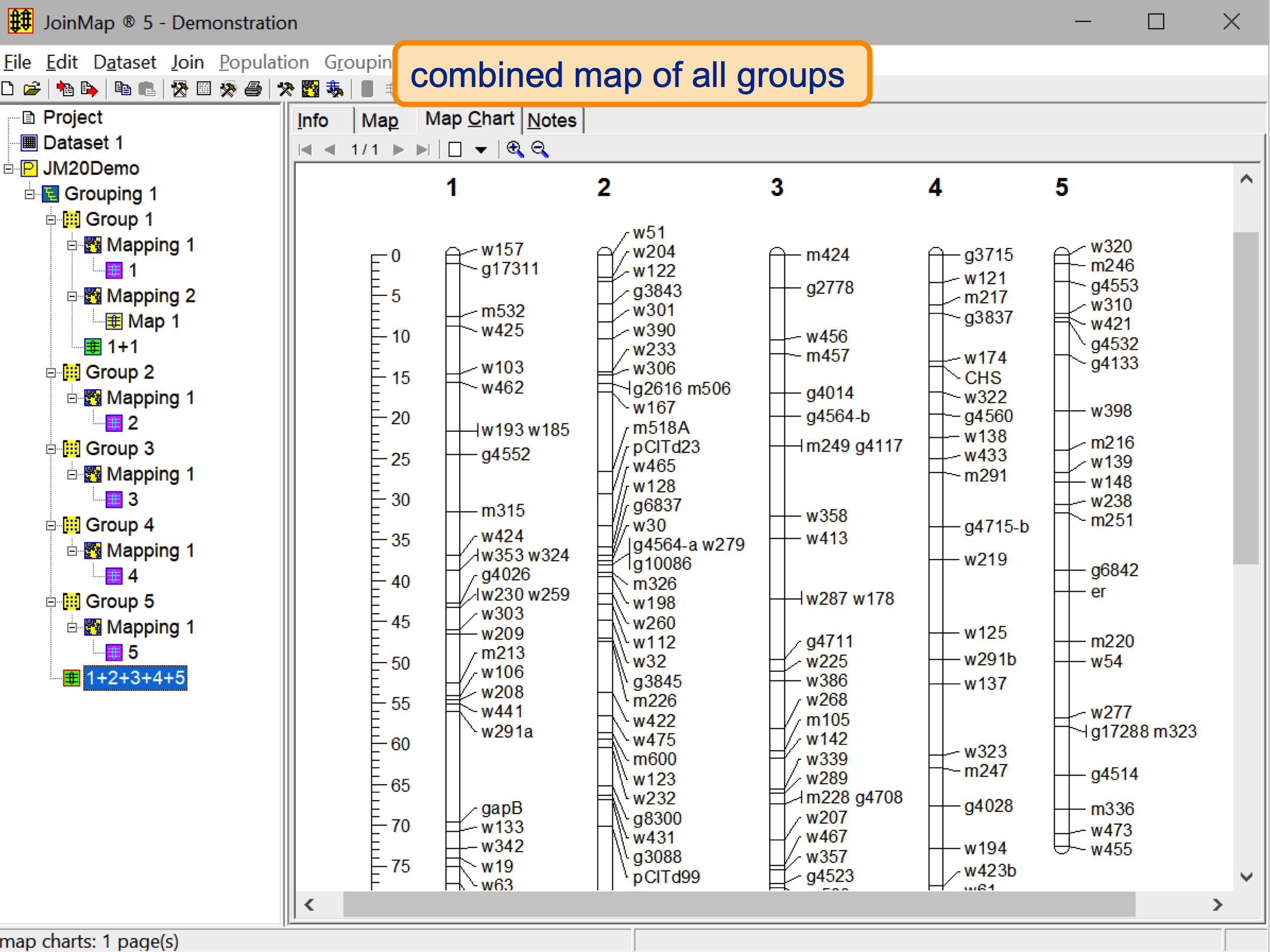
- Combine ('join') data derived from several sources into an integrated map.
- Graphical genotyping.
- No limits to the amount of loci, linkage groups, etcetera, apart from the physical memory (RAM) of the computer.
- Bar and XY charts.
- Map charts with many adjustable features.
- Results and charts exportable to most MS-Windows text processing, presentation and spreadsheet software.
- Export loc-files and maps for use in MapQTL.
For more details: kyazma.nl